This examples propagates an ionized water molecule using the xmolecule electronic structure toolkit.
The dynamics is modelled using fewest switches surface hopping and the electronic structure is calculated using Koopanns’ theorem.
R.log contains the position of the atoms for each time step.
V.log contains the velocity of the atoms.
E.log contains the current potential energy, kinetic energy and total energy of the trajectory.
Switch.log contains information about surface hops
S.log contains information about the current state index
C.log state coefficients
P.log state populations
V_ad.log adiabatic potential energies as a function of time
partial.log shows partial charges (Mulliken charges) for different time steps
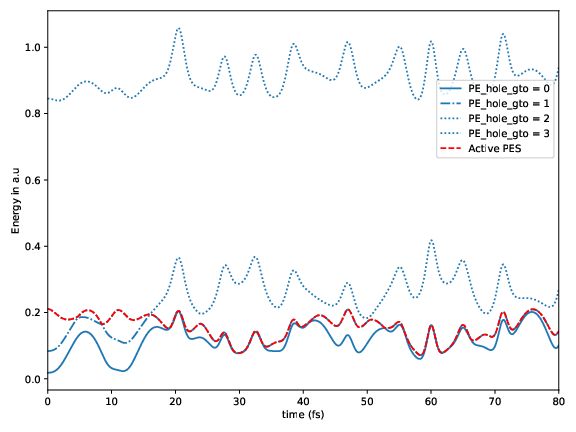
Potential energy for the considered electronic hole states as a function of time for a sample trajectory. The red dash-dotted line marks the active state.
The file input_cis_gto addresses the dynamics of a water molecule in the excited state. The excited state is described using configuratios interaction singles. The following sections are modified compared to the earlier input file:
In the section $xmolecule,
the line CIS=yes switches on configuration interaction singles calculation, nstates=10 indicates that up to 10 states are calculated.
In the section $quantum, the parameter “nstates” was set to 10 (the same number as in the section quantum), and “istate” is set to 3 (the initial state), which means the 3rd excited state (0 means ground state).
Output data for FSSH CIS excited-state trajectory
The folder cis_gto contains the same output files as for the early run for the ionized state.
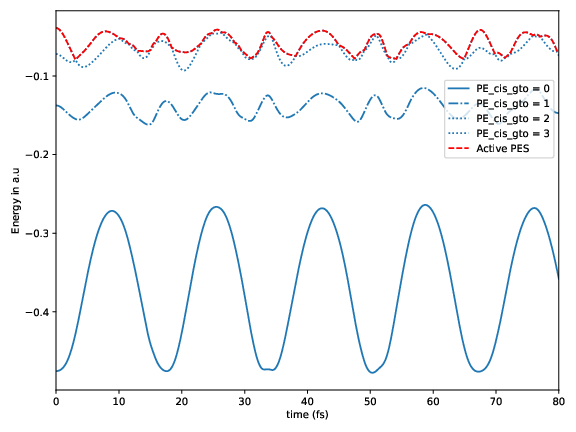
Potential energy for the considered electronic excited states as a function of time for a sample trajectory. The red dash-dotted line marks the active state.
Details of the Inputfile for trivial crossing detection
The file input_cis_olap_gto adds a scheme, in which state overlaps are calculated in each step of the MD calculation. This identifies trivial crossings along the trajectory. In order to do so the option:
trivialCrossing=overlap
is added to the $quantum parameter section.
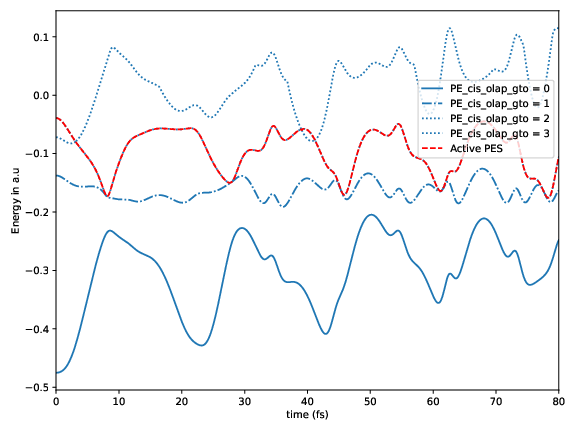
Running the dynamics: detection of trivial crossings
In the folder cis_olap_gto, the additional file V_cross.log contains potential energies as a function of time without trivial crossings. Note that the order of state can change!
The additional file trivialCrossings.log logs the appearance of trivial crossings.
Potential energy for the considered electronic excited states as a function of time for a sample trajectory. The red dash-dotted line marks the active state.